Генетика людини
Генетика людини вивчає особливості спадковості та мінливості виду Homo sapiens sapiens. Усі основні загальні закономірності спадковості, установлені для тварин і рослин, справедливі й для людини. Однак зрозуміло, що генетика людини посідає особливе місце серед інших розділів генетики окремих видів.
Важливість генетики людини зумовлена й тим, що вона із самого початку розвивається не тільки як фундаментальна, але й як клінічна дисципліна. Саме дослідження патологічних варіантів ознак (тобто спадкових хвороб) стали основною базою для вивчення закономірностей спадкування в людини. Нині описано понад 4 тис. різних суто спадкових синдромів, а також доведено роль генетичної етіології у схильності до розповсюджених хвороб (онкозахворювань, серцево-судинних, психічних хвороб тощо). З іншого боку, потреби генетики людини (зокрема медичної) стимулюють розвиток сучасних напрямків загальної генетики та молекулярної біології.
Залежно від проблематики, генетика людини має різні напрямки, які розвинулися в самостійні галузі науки: генетика нормальних ознак людини, медична генетика, фармакогенетика, генетика поведінки та інтелекту, геноміка людини, популяційна генетика людини (у тому числі й геногеографія).
Методи, які використовуються в генетиці людини, принципово не відрізняються від загальноприйнятих для інших об'єктів - це популяційно-статистичний, генеалогічний, близнюковий, цитогенетичний, методи генетики соматичних клітин, молекулярно-біологічні методи. Універсальність генетичних законів дає змогу широко використовувати в генетиці людини модельні генетичні об'єкти (наприклад, для перевірки генотоксичних властивостей хімічних речовин), застосовуючи методи моделювання. Особливості використання даних методик у генетиці людини враховують специфіку людини як генетичного об'єкта.
Зрозуміло, що серед усіх живих організмів людина - найбільш цікавий для нас генетичний об'єкт. Але одночасно людину можна вважати найгіршим об'єктом експериментальної генетики: при вирішенні дослідницьких завдань виникають складні обставини методичного та етичного характеру, які зумовлюють проблематичність людини як об'єкта генетичних досліджень. Розглянемо ці труднощі та шляхи подолання їх.
1. Неможливість експериментальних схрещувань у штучних умовах є цілком зрозумілою, і в першу чергу - з етичних міркувань. Не можна, використавши заздалегідь розроблену схему, відбирати батьків із потрібними генотипами та одержувати й аналізувати їхніх нащадків: люди беруть шлюб вільно, незалежно від генотипу партнера й без будь-якої "дослідницької" мети. Обмеження цієї свободи (шляхом заборони небажаних шлюбів чи сприяння бажаним) було основною ідеєю євгеніки -концепції про "поліпшення" людини біологічним шляхом, в основі якої лежить визнання "генетичних основ" фізичної та психічної нерівноцінності рас, класової нерівності. Спроби такого "поліпшення людської породи" здійснювалися під егідою тоталітарних режимів ХХ століття.
Отже, при генетичному аналізі стосовно людини зникає основа гібридологічного методу - експериментальне схрещування. Існує декілька можливостей для подолання цього "недоліку":
• З великої кількості шлюбних пар генетик може вибрати ті, які його цікавлять, і під час аналізу родоводів зробити висновки стосовно характеру спадкування ознаки.
• Дослідник може проаналізувати спадкування ознак у лабораторних ссавців і зробити попередні висновки щодо можливості спадкування подібних ознак у людини.
• Метод гібридизації соматичних клітин дозволяє в деяких випадках здійснювати генетичний аналіз із використанням культури клітин людини. Аналізуючи гібриди, що утворилися в результаті злиття клітин від двох різних індивідуумів можна робити висновки щодо взаємодії неалельних генів (тест на комплементацію мутацій) і проводити генетичне картування.
2. Пізня статева зрілість та довга тривалість генерацій - для
зміни одного покоління в людини потрібно 20-З0 років, що утруднює аналіз спадкування. Головними методичними підходами, які дозволяють вирішити цю проблему, є вивчення великих популяцій людини, реєстрування ознак протягом значного часу (аналіз родоводів), використання методів генетики соматичних клітин ссавців і людини.
3. Невелика кількість нащадків. Проведення статистичного аналізу розщеплення ознак потребує достатньо великої кількості нащадків від однієї пари батьків (див. розділ З). Людина належить до так званих "малоплідних видів" - за один раз рідко народжується більш ніж одна дитина. Таким чином, проводити аналіз розщеплення ознак на прикладі однієї родини практично неможливо.
Для розв'язання цієї проблеми ведеться пошук багатодітних родин або відбирається потрібна кількість невеликих сімей, де спостерігається ознака, що цікавить дослідника. Існують великі родоводи, де прояв деяких ознак можна простежити протягом декількох генерацій: у великому родоводі кількість нащадків буде достатньою для аналізу.
4. Відсутність чистих ліній і неможливість їхнього отримання є іншою об'єктивною причиною, яка утруднює аналіз спадкування ознак. При аналізі спадкування домінантних ознак генетик не завжди може точно визначити генотип батьків, а змушений робити лише більш-менш імовірні припущення.
5. Велика кількість хромосом (груп зчеплення). Xромосомний набір людини складається з 2З пар хромосом і, відповідно, 24 груп зчеплення: 22 аутосоми та дві статеві хромосоми X і Y. Велика кількість хромосом утруднює їхнє генетичне й цитологічне картування, особливо за допомогою методів класичної генетики. Допомагають розв'язати цю проблему методи гібридизації соматичних клітин людини з соматичними клітинами інших видів ссавців (особливо гризунів) і застосування методів молекулярної цитогенетики, таких як флуоресцентна гібридизація in situ (Fluorescence In Situ Hybridization - FISH - визначення на хромосомі місця гібридизації комплементарного флуоресцентно міченого ДНК-зонда). Однак, ураховуючи велику кількість генів у людини (приблизно 21 тис.), слід відзначити, що питання картування хромосом людини залишається ще досить далеким від свого вирішення, навіть на тлі повного секвенування геному людини.
6. Неможливість створення стандартних ум.ов існування для різних груп індивідуумів значно утруднює вивчення спадкування багатьох ознак людини, особливо тих, що успадковуються полігенно. Навколишнє середовище має величезний вплив на формування низки ознак, однак цей вплив не може змінюватися за бажанням дослідника (харчування, мікроклімат, навчання тощо). Значною проблемою для фахівців при діагностуванні спадкових хвороб є також наявність фенокопій. Цей "недолік" людини як об'єкта генетичних досліджень можна подолати, підбираючи з великої різноманітності людських популяцій групи, схожі за спадковими ознаками і впливом середовища. Для вивчення відносного впливу умов середовища та спадковості на формування ознак використовують близнюковий метод і метод "приймаків" (див. нижче).
До описаних труднощів, з якими стикається генетика людини, слід додати ряд так званих організаційних недоліків, які, на жаль, мають місце в багатьох країнах: незадовільний стан із збереженням документації та реєстрацією смертності, діагностикою і статистикою захворювань.
Незважаючи на перераховані проблеми, людина як генетичний об'єкт має також ряд важливих переваг: існує великий масив даних щодо нормальної та патологічної анатомії, фізіології та біохімії людини; існує можливість спілкування з об'єктом дослідження, що полегшує отримання інформації про його родичів і допомагає досліджувати ознаки, пов'язані з відчуттями, емоціями, інтелектом; практично повністю встановлено геном людини.
Проект розшифрування геному людини (Human Genome Project) був одним із найамбітніших наукових проектів ХХ століття. Він тривав 13 років і закінчився у 2003 р. Проект мав на меті побудувати достатньо точну генетичну карту (з кроком 2-5 сМ), отримати послідовність 3 млрд пар основ ДНК, ідентифікувати генні послідовності, визначити геномні варіації. У результаті виконання проекту побудована генетична карта з кроком у 1 сМ, отримані послідовності 99,9 % еухроматинових ділянок геному з точністю 99,99 %, отримано 15 тис. повних кДНК (змістовних послідовностей білкових генів, див. розділ 9) та ідентифіковано близько З млн нуклеотидів, що варіюють у геномах людини (SNP - single nucleotide polymorphism).
Традиційно геном людини представляють як сукупність послідовностей ДНК 22 аутосом і двох статевих хромосом X та Y. Незважаючи на великий розмір геному (приблизно 3,2 млрд пар основ), верхня оцінка кількості білкових генів у геномі людини не перевищує 25 тис.
(ідентифікація всіх генів РНК є досить далекою від остаточного завершення). Останні версії каталогу білкових генів людини включають близько 21 тис. білкових генів і 4 тис. генів РНК (остання оцінка є напевно заниженою). Загальна довжина кодуючих ділянок генів дорівнює приблизно 34 млн пар основ, тобто становить 1,2 % геному. Складнішим є визначення зв'язку між генним локусом, продуктом і фенотипом, який він може зумовлювати. На 15 жовтня 2008 р. база даних OMIM містила тільки 12 тис. 514 генів, для яких відомі фенотипові прояви.
Щодо мітохондріальної ДНК людини, геном мітохондрій представлений кільцевою молекулою ДНК розміром 16 тис. 569 пар основ і містить 37 генів: гени білків, які беруть участь в окисному фосфорилю-ванні, 2 гени рРНК і 22 гени тРНК.
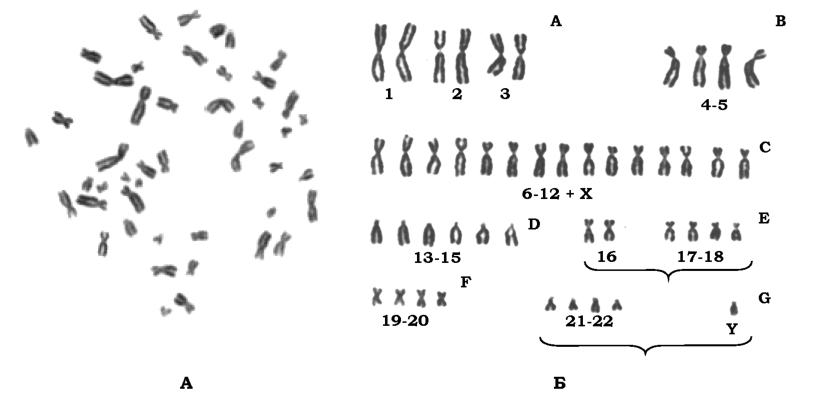
Ядерна геномна ДНК людини, як у інших еукаріотів, організована у хромосоми (див. розділ 1). Нормальний хромосомний набір людини складається із 46 хромосом: 22 пар аутосом і пари статевих хромосом (XX у жінок та XY у чоловіків). Рівномірно пофарбовані метафазні хромосоми людини за розміром і морфологією поділяються на сім груп (позначаються латинськими літерами від A до G, рис. 7.1).
 |
|
Рис. 7.1. Рівномірно пофарбовані метафазні хромосоми людини: (а) - метафазна пластинка; (б) - розкладка хромосом по групах (A-G); окремі хромосоми в межах груп, як правило, неможливо дискримінувати за такого способу фарбування (за винятком хромосом 1, 2, 3, 16 і У) |
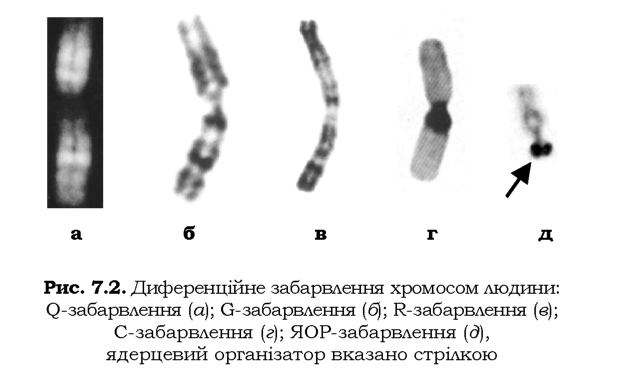
Для більш точної ідентифікації хромосом та їхніх специфічних ділянок застосовують спеціальні методи диференційного забарвлення хромосом, які базуються на використанні певних барвників і процедур попередньої обробки препаратів (рис. 7.2). Найпоширенішим методом диференційного пофарбування є метод G-забарвлення, який дозволяє отримати хромосоми, нерівномірно пофарбовані по довжині. Малюнок чергування темних і світлих смуг є унікальним для кожної пари гомологічних хромосом. Цікаво, що темні та світлі смуги асоційовані з ділянками, збагаченими на AT- і GC-пари відповідно (тобто, відповідно, з геномними зонами, збагаченими на мобільні елементи та кодуючі послідовності). Практично таку саму картину чергування смуг дає метод Q-забарвлення (базується на використанні флуоресцентних барвників). Малюнок сегментів при R-забарвленні є протилежним до такого, що спостерігається при G- і Q-забарвленнях: R-позитивні смуги відповідають GC-збагаченим ділянкам геному. Існують також методи диференційного забарвлення, що дозволяють, наприклад, виявляти центромери та перицентромерний гетерохроматин (С-забарвлення) або ядерцеві організатори - місця локалізації кластерів генів рРНК - (ЯОР-забарвлення), які знаходяться у людини в ділянках вторинної перетяжки коротких плечей акроцентричних хромосом (хромосоми 13-15, 21 і 22).
Результати аналізу геному людини свідчать про надзвичайно високу подібність послідовностей ДНК у різних індивідуумів: відповідно останнім оцінкам, будь-які дві людини є ідентичними за нуклеотид-ними послідовностями на 99,5 %, тобто вся сукупність різноманітних фенотипів у людини зумовлюється варіаціями тільки 0,5 % геному.
Першими, ще задовго до появи методів секвенування ДНК (установлення послідовності, див. розділ 9), були описані варіанти геному людини, які характеризуються зміною кількості й структури хромосом. Анеуплоїдії та хромосомні перебудови часто асоційовані з різними захворюваннями, але гетероморфізм гомологічних хромосом (наявність у них сегментів, які відрізняються за розміром, морфологією або особливостями забарвлення) не обов'язково пов'язаний з патологічними змінами фенотипу й достатньо часто зустрічається в людській популяції (хром.осом.ний поліморфізм). Такі великі геномні зміни, що торкаються послідовностей ДНК розміром понад 3 млн пар основ, визначають як мікроскопічні структурні варіанти.
У процесі аналізу нуклеотидних послідовностей людини були виявлені численні "дрібномасштабні" варіанти геномних послідовностей, розмір яких не перевищує 1 тис. пар основ. Приблизно 80 % таких ДНК-варіантів представлено у вигляді поодиноких нуклеотидних замін (SNP). За приблизними оцінками в людській популяції одна нук-леотидна заміна зустрічається на кожні 300 нуклеотидних пар. Крім SNP до "дрібномасштабних" варіантів геному відносять невеликі делеції, дуплікації та інверсії (див. рис. 4.1), варіації кількості мікро- та мінісателітних повторів. SNP і дрібні перебудови спостерігаються також у мітохондріальному геномі.
В останні три-чотири роки були розроблені нові підходи ефективного аналізу структури геному, які дозволили виявити варіації нукле-отидних послідовностей, що мають проміжну довжину між 1 тис. та 3 млн пар основ. Такі геномні варіанти отримали назву субмікроскопічних структурних варіантів. Субмікроскопічні структурні варіанти в основному представлені інверсіями та CNVs (Copy-Number Variants) - варіантами за кількістю копій елементів послідовності порівняно з "еталонним геномом". Нині описано декілька сотень субмікроскопічних структурних варіантів.
При дослідженні індивідуальних геномних варіацій основну увагу приділяють SNP та іншим "дрібномасштабним" геномним варіаціям. Вважається, що саме вони зумовлюють різноманітність фенотипових варіантів, а також можуть бути відповідальними за схильність до деяких хвороб за дії несприятливих факторів середовища. Очевидно, що більша частина "дрібномасштабних" варіацій спостерігається в некоду-ючих ділянках геному (яких просто значно більше), і такі варіації не впливають на структуру білків. Тому аналіз в основному зосереджують на екзонних варіантах, особливо на несинонімічних нуклеотидних замінах. Проте, накопичуються дані про роль синонімічних замін і замін у некодуючих ділянках у деяких фенотипових проявах (див. розділ 4).
Крім установлення зв'язку поліморфізм - фенотип, "дрібномасштабні" варіації геному використовуються для потреб медичної генетики (при виявлені носіїв рецесивних мутантних генів), популяційної генетики людини (визначення спорідненості між популяціями людей) і судово-медичної експертизи при встановлені особистості або батьківства (у цьому випадку широко використовують поліморфізм за міні- та мік-росателітними локусами). Міні- та мікросателіти є гіперваріабельними тандемними повторами. Їхня різноманітність настільки висока, що кожна людина (крім монозиготних близнюків) має індивідуальний набір варіантів даних послідовностей. За цими наборами одну людину можна відрізнити від іншої так само легко, як і за відбитками пальців, тому методика ідентифікації особистості за міні- та мікросателітними повторами отримала назву ДНК-фінгерпринтингу (див. розділ 9).
Інформація про геном людини, особливо про індивідуальні структурні варіанти геному, розширила наше уявлення про зв'язок генотип -фенотип. Швидкий розвиток методів секвенування ДНК дозволяє говорити про "генетичну паспортизацію" (установлення повних послідовностей геномів кожної людини) як про досить близьку перспективу.
ВИЗНАЧЕННЯ ТИПІВ СПАДКУВАННЯ В ЛЮДИНИ. СКЛАДАННЯ РОДОВОДІВ
Поряд із методами молекулярної генетики, не втратили свого значення традиційні підходи генетичного аналізу людини. Генетичний аналіз будь-якої ознаки в першу чергу має починатися із з'ясування питання, чи успадковується ознака, що нас цікавить. Якщо ознака є спадковою, то другим питанням буде тип її спадкування. Майже універсальним методом, який дозволяє отримати відповіді на дані запитання при дослідженні спадкування ознак людини, був і залишається метод складання та аналізу родоводів. При їхньому складанні ведуться детальні записи про кожного із членів родини та ступінь спорідненості між ними. Далі інформація про родину відображається у графічному вигляді: за допомогою спеціальної символіки (рис. 7.3) будується генеалогічне дерево (педігрі).
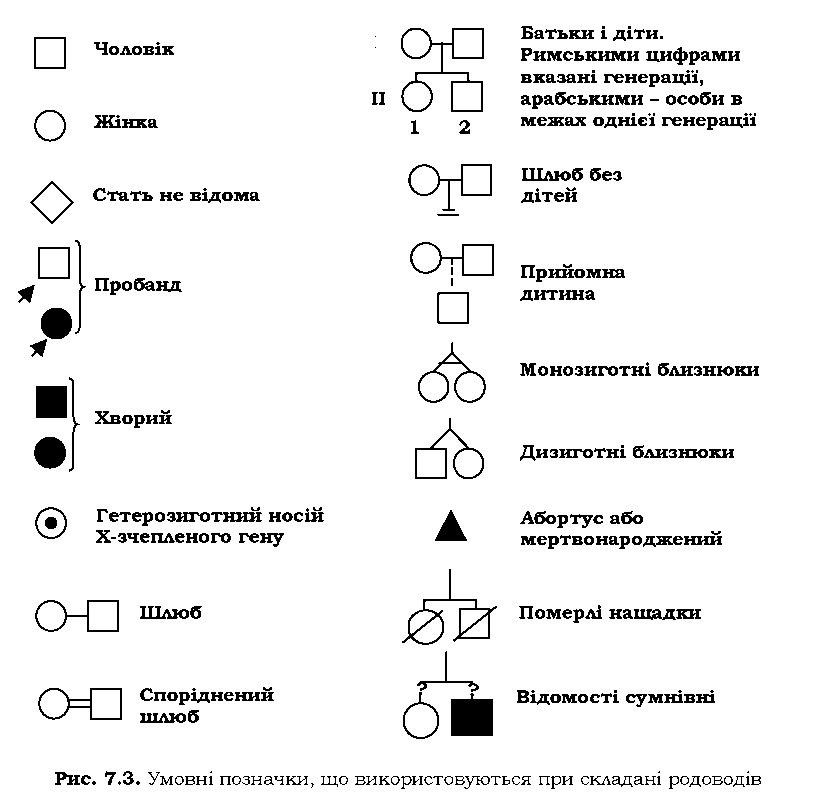
Генеалогічний метод можна застосовувати, якщо є відомості про прямих родичів пробанда - людини, родовід якої складається, -по материнській і батьківській лініях у низці поколінь, та є достатня кількість нащадків у кожному поколінні. В іншому випадку збирають дані щодо достатньої кількості різних родин, де виявляється ознака, яка цікавить дослідника. За отриманими родоводами роблять висновок про можливість спадкування ознаки та тип 'її спадкування (домінантна або рецесивна, аутосомна чи зчеплена зі статтю тощо). Цей формальний аналіз базується на елементарних генетичних закономірностях спадкування (див. розділ 3).
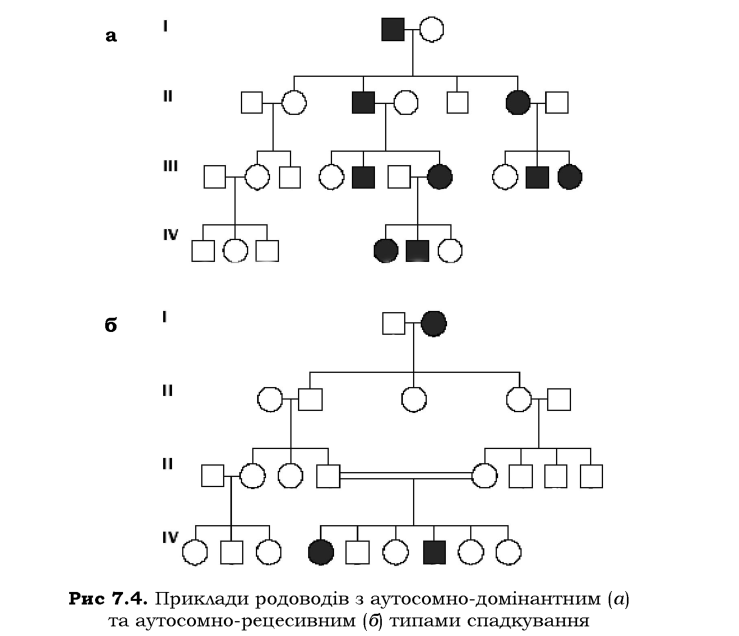
Для родоводу, де спостерігається аутосомно-домінантний тип спадкування ознаки (приклад генеалогічного дерева див. на рис. 7.4, а), є характерною наявність носіїв ознаки в кожному поколінні - "вертикальне" спадкування. Якщо в батьків ознака не проявляється, імовірність народження дитини з такою ознакою незначна (у протилежному випадку можна говорити про виникнення генеративної мутації, див. розділ 4). Ознака виявляється з однаковою частотою серед чоловіків і жінок: показник того, що ген, який зумовлює ознаку, міститься в аутосомі.
Зрозуміло, що остання закономірність зберігається і для аутосом-но-рецесивного типу спадкування (рис. 7.4, б), яке в інших відношеннях буде "негативом" до родоводу з аутосомно-домінантним типом. Зокрема, у родоводі наявні одне чи декілька поколінь, в яких прояв ознаки не спостерігається. Якщо ж ознака виявляється, то вона часто представлена в більшості рідних або двоюрідних сибсів (братів і сестер) - "горизонтальне" спадкування. У випадку, коли у здорових батьків (домінантна ознака) народжується хвора дитина (рецесивна ознака), батьки дуже часто є родичами.
Приклади деяких домінантних і рецесивних ознак людини наведено в табл.7.1.
|
Таблиця 7.1. Деякі домінантні та рецесивні ознаки в людини |
||||||||||||||||||||||||||||||||||||||||||||||||||
|
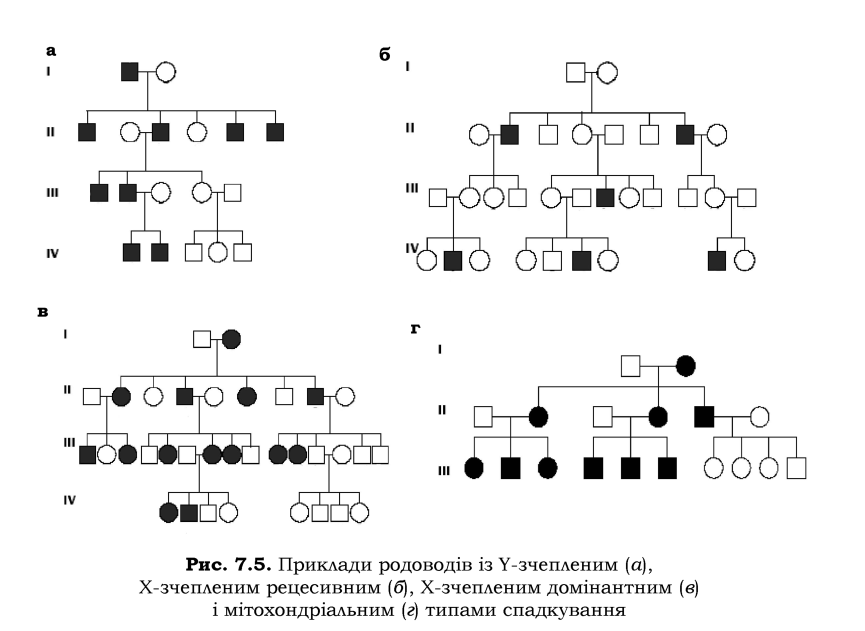
Не викликає труднощів аналіз родоводів, якщо ознака зчеплена зі статтю (рис 7.5, а-в). Деякі ознаки, зчеплені зі статтю в людини, наведено в табл. 7.2 (слід нагадати, що псевдоаутосомні ознаки (див. розділ 6) успадковуються аналогічно тим, гени яких знаходяться в аутосомах).
|
Таблиця 7.2. Деякі ознаки, зчеплені зі статтю в людини |
||||||||||||||||||||
|
Найпростіше можна виявити голандричні ознаки (зчеплені з Y-хромосомою): ознака буде проявлятися виключно у чоловіків і передаватися від батька до сина зі 100 % імовірністю (рис. 7.5, а).
При X-зчепленому рецесивному типі успадкування (рис. 7.5, б) прояв ознаки буде зустрічатися майже виключно у чоловіків, ознака ніколи не буде передаватися від батька до сина, але тільки до онука (чи правнука) через доньку.
Х-зчеплений домінантний тип успадкування при аналізі педігрі можна сплутати з аутосомно-домінантним типом. Основними характеристиками, що вказують на зчеплення з X-хромосомою, є різна частота прояву ознаки серед чоловіків і жінок - серед жінок вона зустрічається частіше - та те, що ознаку батька успадковують всі доньки і ніколи - сини (рис. 7.5, в).
Деякі ознаки можуть бути зумовлені мітохондріальними генами. Як відомо, дітям передаються виключно мітохондрії матері, у результаті чого при спадкуванні ознак спостерігатиметься материнський ефект: чоловіки та жінки можуть мати відповідну ознаку, але тільки жінки передають її своїм дітям (рис. 7.5, г). У людини як мітохондріальні успадковуються, наприклад, оптична атрофія Лебера (втрата зору в центральній частини зорового поля) та онкоцитома (доброякісна пухлина нирок).
Генеалогічний метод дозволяє визначити не тільки розглянуті типи спадкування. Аналіз складніших випадків (наприклад, коли ознака має неповну пенетрантність або різну експресивність, є залежною від статі або обмеженою статтю) також не викликає принципових труднощів.
Крім визначення типів спадкування, аналіз родоводів за декількома ознаками використовується для встановлення зчеплення генів і розрахунку відстані між ними. За наявності великого родоводу алгоритм виявлення факту зчеплення та розрахунку відстані між генами є майже подібним до описаного в розділі 3. Проте великі родоводи на практиці зустрічаються достатньо рідко. Найчастіше мають справу з великою кількістю маленьких родоводів, які аналізують за допомогою спеціальних комп'ютерних програм.
КІЛЬКІСНІ Й БАГАТОФАКТОРНІ ОЗНАКИ ЛЮДИНИ
Найбільш вивченими є закономірності успадкування якісних моно-генних ознак. Але, як згадувалося в розділах 3 і 4, гени не функціонують в "ізоляції" від інших генів та навколишнього середовища. Прояв великої кількості ознак людини (як і інших організмів) зумовлюється одночасно багатьма факторами: дією декількох генів та впливом комплексу чинників довкілля (кількісні або багатофакторні ознаки).
Найпростішим типом спадкування кількісних ознак є полімерія, коли прояв ознаки залежить від наявності в генотипі особини відповідної кількості домінантних алелів однозначних генів (декількох копій одного гена), що діють адитивно (розділ 3). Класичним прикладом у людини є спадкування кольору шкіри, який, за підрахунками, визначається п'ятьма генами. Однак для інших кількісних ознак (зріст, маса тіла тощо) ситуація є значно складнішою. Гени, що відповідають за дані ознаки, хоча й діють на перший погляд адитивно (роблять свій внесок у неперервний розподіл варіантів ознаки), але не є однозначними, і між ними можуть відбуватися взаємодії різного типу.
Багатофакторні ознаки можуть виявлятися як якісні, коли особа або має ознаку, або ні. Наприклад, деякі уроджені вади розвитку (аненцефалія, спинномозкова грижа) і більшість розповсюджених захворювань (інфаркт міокарда, астма, атеросклероз, "спорадичні" он-козахворювання тощо) є якісними ознаками, але закономірності їхнього успадкування такі самі, як у кількісних. Для пояснення розподілу подібних ознак у популяціях людей була запропонована модель порогу: ознака проявляється тільки у тих особин, у яких кількість "генів схильності" перевищує певний "поріг", тобто кількісною ознакою є схильність до хвороби, а не симптоми захворювання. У медичній практиці модель порогу застосовують також і для "класичних" кількісних ознак, об'єднуючи відповідні варіанти в так звану "норму" й "патологію" - це, наприклад, дистрофія, нормальна вага, клінічне ожиріння. Величина "порогу" для окремих індивідуумів залежить від різноманітних чинників - статі особи, способу життя, факторів довкілля. Умови життя можуть значно збільшувати чи зменшувати кількість "генів схильності", необхідну для прояву ознаки та, відповідно, впливати на ризик розвитку захворювання.
Близнюковий метод і метод приймаків
Значна залежність прояву багатофакторних ознак від зовнішніх чинників утруднює їхній генетичний аналіз і пошук генів, що їх обумовлюють. Навіть у межах однієї родини інколи важко з'ясувати, чи схожість за деякими ознаками між родичами підтверджує спадковість цієї ознаки, чи свідчить тільки про ідентичність умов існування. Для відокремлення дії факторів зовнішнього середовища від впливу генотипу на прояв ознаки в генетиці людини використовують близ-нюковий метод і метод приймаків.
Близнюковий метод базується на порівнянні збігу прояву ознак у близнюків. У людини близнюки народжуються з частотою приблизно 1 двійня на 84 пологи (трійні - 1 на 7 тис.). Одну третину з них становлять м.онозиготні або однояйцеві близнюки Вони розвиваються з бластомерів однієї заплідненої яйцеклітини, які розділилися на початкових стадіях дроблення (найчастіше на стадії двох бластомерів) і дають початок окремим організмам. Отже, монозиготні близнюки мають ідентичні генотипи. А дизиготні розвиваються з двох яйцеклітин, які дозріли й були запліднені одночасно. За генотипом вони схожі не більше, ніж рідні брати й сестри, що народилися в різний час.
При обстеженні одностатевих близнюкових пар важливим є точне встановлення типу їхньої зиготності. Його визначають або за подібністю прояву в пари близнюків ознак, що перебувають у мінімальній залежності від середовищних факторів (групи крові за різними антигенами, колір очей, можливість відчувати смак ФТК тощо), або використовуючи ДНК-фінгерпринтинг. Ознаки, прояв яких збігається в пари близнюків, називають конкордантними (якщо прояв відсутній у одного з них - дискордантними).
Вплив спадковості на прояв ознаки розраховують, порівнюючи конкордантність ознак (відсоток пар близнюків з ідентичним проявом потрібної ознаки) у монозиготних і дизиготних близнюків (таблиця 7.3). Зрозуміло, що, оскільки генотипи в монозиготних близнюків однакові, то фенотипові відміни між ними будуть зумовлені виключно впливом середовища під час внутрішньоутробного розвитку та після народження. Що стосується збігу в прояві ознак, то він може пояснюватися як ідентичністю генотипів, так і подібністю умов проживання. У цьому аспекті цікавими є випадки, коли пару монозиготних близнюків розлучали в дитинстві й тривалий час вони виховувались і росли в різних умовах. Конкордантність у таких пар поясню-

|
Ознака |
Конкордантність, % |
|
|
МБ |
ДБ |
|
|
Група крові (АВ0) |
100 |
46 |
|
Колір очей |
99 |
28 |
|
Колір волосся |
89 |
22 |
|
Розумова відсталість |
97 |
37 |
|
Підвищена вага тіла у віці 18-20 років |
51,8 |
20,2 |
|
Підвищена вага тіла у віці після 45 років |
53 |
27,4 |
|
Епілепсія |
72 |
15 |
|
Туберкульоз |
56 |
22 |
|
"Заяча губа" |
42 |
5 |
|
Кір |
98 |
94 |
|
Шизофренія |
46 |
14 |
|
Маніакально-депресивний психоз |
60 |
14 |
Чисельно вклад генотипу в розвиток тієї чи іншої ознаки виражається як коефіцієнт спадковості H, який у даному випадку можна розрахувати за формулою
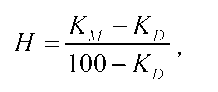
де Km - конкордатність (у відсотках) монозиготних, Kd - дизиготних близнюків. Якщо H = 1, то ознака повністю визначається генотипом, при H = 0 - факторами середовища. Наприклад, коефіцієнт спадковості для кольору волосся дорівнює 0,86, що говорить про переважний внесок генетичних факторів у формування ознаки, але існує також незначний вплив середовища (0,14). Для надлишкової ваги тіла коефіцієнти спадковості дорівнюють 0,4 (для віку 18-20 років) і 0,35 (для людей після 45 років), що вказує на значніший вплив середовищних факторів, але спадковий компонент також відіграє суттєву роль.
Іншім підходом до оцінки ролі генотипу й середовища в розвитку багатофакторних ознак є аналіз фенотипових проявів у дітей, які були прийняті в родину - метод приймаків. Приймаки та прийомні ро-
дичі мають абсолютно несхожі генотипи, але подібні умови існування. Найчастіше метод приймаків використовується в комплексі з близ-нюковим, що дає можливість отримати більш об'єктивні результати щодо генетичного впливу на розвиток кількісних ознак.
Дослідження типів спадкування ознак і пошук генів, які зумовлюють чи модифікують їхній прояв, мають велике практичне значення, коли йдеться про патологічні прояви ознак. Хоча патологічні ознаки є крайніми варіантами прояву нормальних ознак, існує окремий розділ генетики людини, який зосереджує увагу на ролі спадковості у розвитку саме патологічних станів або хвороб - медична генетика. Залежно від внеску спадкових і середовищних факторів у розвиток патологічних станів і перебіг захворювань, усі хвороби можна умовно поділити на три групи: спадкові (незначний внесок середовища в розвиток патології), хвороби зі спадковою схильністю (розвиток патології зумовлюється взаємодією генотипу й середовища) і неспадкові захворювання (патології, викликані зовнішніми факторами).
Медична генетика спрямована на діагностування, лікування, прогнозування та профілактику спадкових захворювань, у тому числі хвороб із спадковою схильністю, до яких відносять більшість захворювань людини. У прояві патології важливу роль відіграє комбінація спадкових і середовищних факторів, тобто хвороби є багатофактор-ними ознаками. Ефективна (у перспективі) діагностика схильності до таких хвороб і оцінка індивідуальних ризиків тісно пов'язана з розвитком методів секвенування індивідуальних геномів.
Суто спадковими захворюваннями називаються хвороби, причинами яких є мутації - зміни спадкового апарату. Хвороби, викликані точковими мутаціями, належать до генних спадкових захворювань. Захворювання, які спричинені зміною структури та кількості хромосом, об'єднують у групу хром.осомних хвороб. Слід зауважити, що спадкові хвороби не обов'язково передаються наступним поколінням. Так, основна частина хромосомних хвороб не успадковується (унаслідок патологій репродуктивної системи хворих). Більшість онкологічних хвороб зумовлена виникненням соматичних мутацій і також не успадковується наступними поколіннями.
Більша частина спадкових синдромів викликана різноманітними патологічними генними мутаціями. Такі мутації характеризуються плейот-ропною дією (один синдром - сукупність симптомів) і високою пенетран-тністю, їхні прояви майже не залежать від факторів навколишнього середовища. Патологічна симптоматика може бути спричинена відсутністю продукту пошкодженого гена, зменшенням або збільшенням кількості генного продукту, а також утворенням аномального продукту. Генні хвороби класифікують за фенотиповими проявами й типом спадкування. При класифікації за фенотипом за основу беруть або системні симптоми (спадкові хвороби нирок, опорно-рухової системи тощо), або біохімічні прояви (порушення обміну речовин, аномалії структурних білків).
Серед спадкових вад метаболізму виділяють хвороби, пов'язані з порушенням обміну амінокислот (аміноацидопатіі), вуглеводів, ліпідів, нуклеїнових кислот і мінеральних речовин. Патологічні прояви можуть бути зумовлені мутаціями генів, що кодують ферменти (ензимопатіі); білки, які регулюють активність генів ферментів чи активність самих ферментів; білки, які забезпечують транспорт необхідних для метаболізму речовин; рецептори тощо. Метаболізм будь-яких речовин є процесом багатоступеневим, тому мутації різних генів, які забезпечують перетворення речовин від етапу проникнення речовини в організм до безпосередньої дії метаболітів на відповідні клітини, можуть приводити до подібних патологічних фенотипових проявів. У даному випадку говорять про генетичну гетерогенність спадкових захворювань, а мутації різних генів, які викликають схожу клінічну картину, називають генокопіями.
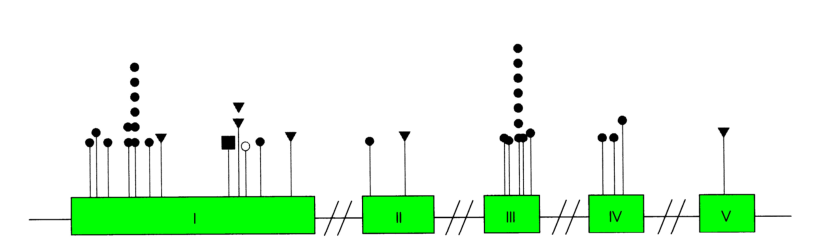
Прикладом спадкового синдрому порушення метаболізму амінокислот є альбінізм, причиною якого є мутація в гені тирозинази (розташований у довгому плечі хромосоми 11) - ферменту, що перетворює тирозин на дигідроксифенілаланін (ДОФА), який є субстратом для синтезу меланіну. У результаті захворювання характеризується відсутністю пігментації: хворі мають молочно-білий колір шкіри й біле волосся. Захворювання зустрічається з частотою 1/28-39 тис. і успадковується за аутосомно-рецесивним типом. Наразі відомо близько 40 мутацій у гені тирозинази, які приводять до альбінізму (деякі наведено на рис. 7.6). Мутації в різних частинах гена можуть зумовлювати варіанти фенотипового прояву хвороби. Так, мутація в кодоні 81 (пролін замінюється на лейцин) приводить до класичного типу альбінізму (позначається як IA), мутація в кодоні 406 (також заміна проліну на лейцин) є причиною прояву альбінізму типу IB. Тип IB, або "жовтий мутант", відрізняється від класичного типу наявністю у хворих волосся жовтого кольору та значно меншою чутливістю шкіри до сонячних променів.
 |
|
Рис. 7.6. Мутації в гені тирозинази. Римськими цифрами позначено екзони, колами - однонуклеотидні заміни, трикутниками - мікроделеції, квадратом - інсерція |
Іншою аміноацидопатією, пов'язаною з порушенням обміну тирозину, є фенілкетонурія (ФКУ). Хвороба зумовлена мутаціями в гені фенілаланінгідроксилази (усього відомо близько 600 мутацій). Через відсутність активного ферменту фенілаланін не перетворюється на тирозин. Причиною патології є фенілаланін, токсичний за великих концентрацій, а також його токсичний метаболіт фенілпіровиноград-на кислота. Хворі при народжені мають специфічний "мишачий" запах, дисморфічне обличчя та недостатність пігментації (світла шкіра й волосся). Пізніше з'являються судоми, розвивається розумова відсталість. Паталогічних проявів мутацій можна уникнути за умови, що одразу після народження й до 15 років дитина буде переведена на спеціальну безфенілаланінову дієту. ФКУ зустрічається з достатньо високою частотою - у середньому 1/10 тис. (у деяких популяціях в Україні частота досягає 1/4,5 тис.), спадкується як аутосомно-рецесивна ознака. Цікавим є те, що кількість гетерозигот за мутантним геном фенілаланінгідролази в популяції є дещо більшою, ніж можна встановити за формулою Харді - Вайнберга (див. розділ 8). Це пояснюється тим, що підвищений рівень фенілаланіну в гетерозиготних жінок є фактором, який зменшує ризик викиднів.
Нестача ферменту може привести до поступового накопичення певних токсичних субстратів у клітині (саме у клітині - на відміну від ФКУ). До таких хвороб накопичення відносять, наприклад, хворобу Тея - Сакса (амавротична ідіотія). Через відсутність ферменту гексо-зоамінідази А в лізосомах нервових клітин не розщеплюється гангліо-зид GM2 - один із компонентів клітинної мембрани. Гангліозид накопичується в лізосомах, що веде до загибелі нервових клітин. Симптоми хвороби починають виявлятися в основному через декілька місяців після народження: спостерігається гальмування розвитку та прогресуючі нервові розлади. Смерть настає в середньому у віці трьох років. Є доросла форма амавротичної ідіотії - хвороба починає розвиватися у віці від 20 до 40 років і супроводжується поступовою прогресуючою розумовою відсталістю. Дитяча форма викликана мутаціями в гені гексозоамінідази А за типом зсуву рамки зчитування або порушення сайтів сплайсингу, доросла - місенс-мутаціями.
Муковісцидоз є прикладом захворювання, яке пов'язане з порушенням обміну неорганічних іонів. Унаслідок мутації в гені CFRT (картований у довгому плечі хромосоми 7, кодує білок каналу для іона хлору) у хворих спостерігаються дефекти потових та інших залоз, що виникають у результаті дії великої концентрації солі в секретах, а також порушення засвоєння їжі, спричинене блокуванням протоків підшлункової залози. Відповідно, спостерігається затримка росту й розвитку дитини. Найфатальнішим є накопичення слизу в дихальних шляхах, що викликає легеневу недостатність у хворих і провокує розвиток пневмоній. Хвороба (аутососомно-рецесивний тип спадкування) зустрічається з частотою від 1/2 тис. до 1/90 тис.
Порушення транспорту кисню гемоглобіном є причиною патологічних проявів гемоглобінопатій. Хвороби цього класу пов'язані з мутаціями в генах а- або в-субодиниць гемоглобіну. Серпоподібноклітинна анемія зумовлена мутацією, яка викликає заміну 6-ї амінокислоти в ланцюзі в-глобіну (глутамін замінюється на валін). Варіант гемоглобіну (гемоглобін S), до складу якого входить змінений в-глобін, гірше виконує свою функцію. Крім того, він здатен полімеризуватися, утворюючи філаменти, які змінюють морфологію еритроцитів (вони набувають форму серпа - звідти назва хвороби). Гомозиготи за мутантним геном мають широкий спектр симптомів: еритроцити серпоподібної форми, анемію, ниркову, серцеву та легеневу недостатність, розумову відсталість, паралічі, болі в суглобах і животі. Без спеціального лікарського нагляду хворі помирають у ранньому дитинстві. У гетерозигот еритроцити мають неправильну форму, оскільки містять два варіанти гемоглобіну - нормальний і мутантний. Але клінічні прояви захворювання в гетерозигот відсутні (починають виявлятися лише за умови зниження концентрації кисню в повітрі - наприклад, у високогірних умовах). Тобто серпоподібноклітинна анемія успадковується як ознака з неповним домінуванням. Хоча гемоглобін S є патологічним варіантом білка, його ген достатньо розповсюджений, особливо у країнах із великим рівнем захворюваності на малярію: кількість носіїв гена досягає 40 % у деяких країнах східної Африки. Це пояснюється високою резистентністю гетерозигот до малярії.
Іншим типом гемоглобінопатій є таласемії - хвороби, асоційовані з порушеннями експресії а- або в-глобінів (відповідно, розрізняють а- і в-таласемії). До патологій приводять або делеції у глобінових генах, або мутації, що спричиняють зменшення кількості активних мРНК у клітині. Хвороби поширені в деяких середземноморських регіонах: частота хворих може доходити до 30 % (назва хвороби походить від грецького слова таХааа - море). Таласемії характеризуються гемолітичною анемією, жовтухою, гіперплазією кісткового мозку та кістковими аномаліями. Залежно від генотипу хворого, симптоматика варіює від субклінічної (м'яка форма) до важкої летальної. Слід звернути увагу, що серпоподібноклітинна анемія та в-таласемія є прикладом, коли різні мутації одного гена можуть мати зовсім різні фенотипові прояви.
Мутації в генах структурних білків тканин, які призводять до відсутності, нестачі або утворення аномального білка, є також причиною великої кількості спадкових синдромів. Наприклад, синдром Марфана зумовлений мутаціями в гені фібриліну (локалізований у довгому плечі 15-ї хромосоми) - одного з білків сполучної тканини. Хворі мають порушення кістяка, високий зріст, арахнодактилію (павучі пальці) і дефекти серцево-судинної системи (найнебезпечнішим серед симптомів є ослаблення стінок аорти). Синдром Марфана - аутосомно-домінантне захворювання, яке зустрічається з частотою приблизно 1/10 тис. Цікаво, що приблизно 25 % хворих народжується в родинах, де захворювання раніше не спостерігалося, що вказує на високий рівень мутацій гену фібриліну.
В окрему групу спадкових генних захворювань виділяють так звані хвороби експансії тринуклеотидних повторів. Причиною цих хвороб (таблиця 7.4), які не мають спільної симптоматики, є багатократне збільшення числа копій (експансія, див. розділ 4) тринуклео-тидного повтору в межах певного гена. У більшості випадків ці хвороби успадковуються як домінантні ознаки, що виявляються у статевозрілому віці. Якщо у клінічно здорових батьків народжується хвора дитина, то один із батьків мав кількість повторів, більшу за норму, але меншу за потрібну для клінічних проявів (так званий стан прему-тації): при достатній кількості повторів значно зростає імовірність їхньої подальшої експансії при кожному наступному раунді реплікації. Відповідно, для всіх хвороб даного типу характерне явище антиципації - з кожним наступним поколінням симптоматика хвороби посилюється, хвороба виявляється в більш ранньому віці, що пов'язано зі збільшенням кількості копій тринуклеотидного повтору.
|
Захво рювання |
Симпто матика |
Спадку вання |
Одиниця повтору |
Кількість копій |
|
|
Норма |
Патологія |
||||
|
Міотонічна дистрофія |
Прогресуюча м'язова слабкість, судоми, катаракта, серцева аритмія |
Аутосомно- домінантне |
CTG |
5-З7 |
80-З000 |
|
Xорея Геннтинг- тона |
Прогресуюча дегенерація базальних гангліїв, яка супроводжується розладами руху, змінами психіки, епілептичними нападами |
Аутосомно- домінантне |
CAG |
6-З4 |
40-121 |
|
Спинно-церебеляр-на атаксія першого типу |
Прогресуюча дегенерація ділянок мозочка та спинного мозку з поступовою втратою координації рухів |
Аутосомно- домінантне |
CAG |
6-З0 |
41-81 |
|
Атаксія Фрідрейха |
Прогресуюча дегенерація задніх і бокових стовпів спинного мозку з поступовою втратою координації рухів |
Аутосомно- рецесивне |
GAA |
10-21 |
200-900 |
|
Синдром Мартіна -Белла |
Ламка X-хромосома, розумова відсталість |
Зчеплене з X-хромосомою, неповне домінування (ознаки розумової відсталості спостерігаються у 60 % гетерозиготних жінок) |
CGG |
6-52 |
2З0-2000 |
Велика група спадкових патологій належить до хромосомних хвороб. Різноманітні порушення каріотипу зустрічаються серед новонароджених у середньому з частотою 0,6 %. Тільки 10 % від цих аномалій не супроводжується видимими патологічними станами. Але реальний рівень утворення хромосомних аномалій значно вищий: за деякими розрахунками приблизно 25 % зигот мають аномальний каріотип і основна маса ембріонів із такими порушеннями гине ще в доімплатаційний період. Серед спонтанних абортусів 50 % мають хромосомні аномалії.
Порушення в кількості або структурі хромосом можуть виникати при гаметогенезі в одного з батьків. У цьому разі аномалія буде зустрічатися в усіх клітинах зародку. Такий організм називають повним м.у-тантом Інколи хромосомні аномалії виникають у процесі ембріонального розвитку. У результаті тільки частина клітин зародку буде мати аномальний каріотип - таке явище назвали генетичним м,озащизм,ом.
Кількісні аномалії хромосом виникають унаслідок порушення сегрегації хромосом (див. розділ 4). Єдиною сумісною з життям моносо-мією в людини є моносомія X-хромосоми. Моносомії по всіх аутосомах є летальними: зародок гине на початкових стадіях розвитку, оскільки навіть серед абортусів дана аномалія каріотипу не зустрічається. Трисомії по більшості аутосом також спричиняють аномалії розвитку зародку, які несумісні з життям. Серед живонароджених спостерігаються трисомії тільки по хромосомах 21, 18 і 13 (рис.7.7).
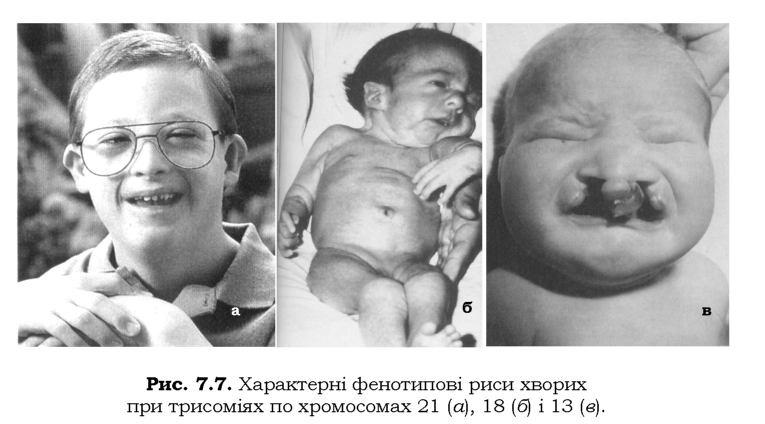
Трисомія-21 - синдром Дауна - найбільш розповсюджена хромосомна хвороба і зустрічається в середньому з частотою 1/500-700 новонароджених. Співвідношення статей серед хворих 1 : 1. Хворі мають характерний фенотип: монголоїдний розріз очей, наявність епіканта, плоске широке перенісся, низький зріст, маленький череп зі згладженою потилицею. У багатьох хворих спостерігаються аномалії серцево-судинної системи та імунодефіцити, які є основними причинами їхньої загибелі. Чоловіки стерильні, однак жінки іноді можуть мати дітей. Одним із головних симптомів синдрому Дауна є розумова відсталість (дебілізм різного ступеня, максимальний IQ = 75). Мозаїчні форми синдрому Дауна (близько 3 % від загальної кількості випадків) характеризуються значно м'якішою симптоматикою. У деяких випадках один із батьків хворого на синдром Дауна (частіше мати) є носієм збалансованої робертсонівської транслокації (див. розділ 4) хромосоми 21 із хромосомами 13-15, рідше з хромосомою 22. У такій родині можуть народжуватися хворі діти із синдромом Дауна, нормальні, а також здорові діти -носії збалансованої робертсонівської транслокації.
Трисомія-18 - синдром Едвардса - зустрічається з частотою 1/4,5-7 тис. новонароджених. Діти при народженні мають маленьку вагу, слабкі, повільно розвиваються фізично та психічно. Найчастіше спостерігаються аномалії черепа (скошене підборіддя, маленький рот, слабко розвинуті щелепи, випнута потилиця), деформовані низько розташовані вуха, деформації рук і стоп, коротка грудина, дефекти розвитку м'язів. Хворі мають значні пороки розвитку серця та нирок. Тільки 1-2 % дітей доживає до 1 року. Особливістю синдрому Едвардса є вища народжуваність дівчат із патологією, ніж хлопчиків (3 : 1), що відображає вищу внутрішньоутробну смертність чоловічих плодів.
Трисомія-13 - синдром Патау. Для клінічної картини захворювання характерними є множинні вроджені вади розвитку: розщеплення м'якого і твердого піднебіння, "заяча" губа, мікрофтальмія (недорозвинені маленькі очі), інколи відсутність очей, вуха, мікроцефалія, затримка росту та розумового розвитку, порушення серця, нирок та травної системи. Синдром зустрічається з частотою 1/14,5 тис., більшість плодів гине протягом внутрішньоутробного розвитку. Діти часто народжуються передчасно й мають маленьку масу, рідко доживають до 1 року, ті, що вижили, страждають глибокою формою ідіотії. Описано випадки, коли причиною синдрому Патау були робертсонівські транслокації за участю хромосоми 13. При мозаїцизмі за додатковою хромосомою 13 часто спостерігають розумову відсталість без зовнішніх аномалій.
Кількісні аномалії статевих хромосом представлені моносомією хромосоми X і полісоміями X- та Y-хромосом. Основні синдроми, причиною яких є зміна нормального числа статевих хромосом, наведено в табл. 7.5.
|
Таблиця 7.5. Основні синдроми, пов'язані зі зміною кількості статевих хромосом |
||||||||||||||||||||
|
синдрому Кляйнфельтера. Для всіх трисомій із материнським ефектом є характерною чітка позитивна кореляція між віком матері та ризиком розвитку плоду з відповідним аномальним каріотипом. Збільшувати ризик нерозходження хромосом у матері можуть також зовнішні фактори: інфекційні хвороби, мутагенні чинники та шкідливі звички (тютюнопаління, алкоголізм тощо).
Описані кількісні аномалії хромосом - це анеуплоїдії. Причиною 2З % спонтанних абортів є поліплоїдії. Серед живонароджених тетраплої-дів не спостерігається, є одиничні випадки народження триплоїдних дітей із множинними важкими вадами розвитку, не сумісними з життям.
Крім кількісних аномалій хромосом, причиною вроджених патологічних станів можуть бути структурні хромосомні аберації. Такі хромосомні хвороби поділяються на синдроми часткових анеуплоїдій (до цієї групи не відносять незбалансовані робертсонівські транслока-ції) і мікроцитогенетичні синдроми.
Синдроми часткових анеуплоїдій (на сьогодні їх описано близько 100) отримали свою назву тому, що патологічна симптоматика захворювань зумовлена або надлишком (часткова трисомія), або нестачею (часткова моносомія) досить великої ділянки певної хромосоми. Основна клінічна картина, як і для повних анеуплоїдій, крім комбінації притаманних даному синдрому ознак, характеризується великим набором неспецифічних симптомів: уроджені вади розвитку, розумова відсталість, дисплазії, дефекти серцево-судинної системи тощо. Неспецифічність симптоматики вказує на те, що в загальній патології основну роль відіграють не втрачені чи надлишкові генні локуси, а сам факт хромосомного дисбалансу.
До мікроцитогенетичних синдромів відносять захворювання, причиню яких є субмікроскопічні делеції або дуплікації відповідних ділянок хромосом. Мікроцитогенетичні синдроми зустрічаються з достатньо низькою частотою (1/50 тис. - 1/100 тис.), характеризуються чіткою клінічною картиною. Прикладами мікроцитогенетичних синдромів є спадкова ретинобластома (мікроделеція в хромосомі 13, яка є причиною вроджених злоякісних пухлин сітківки ока), синдром Бек-віта - Відеманна (мікродуплікація в короткому плечі хромосоми 11, яка приводить до характерних вад розвитку: грижа пупкового канатика, мікроцефалія, гіпоглікемія та множинні пороки розвитку внутрішніх органів), синдроми Ангельмана та Прадера - Віллі (мікроделе-ції в довгому плечі хромосоми 15).
З перерахованих захворювань синдроми Ангельмана та Прадера -Віллі привертають особливу увагу. При синдромі Прадера - Віллі хворі мають неконтрольований апетит і, як результат, ожиріння, диморфізм обличчя, маленькі стопи й долоні, гіпотонію, розумову відсталість. Синдром Ангельмана характеризується важкими нервовими, психічними та розумовими розладами: порушення координації рухів (маріонеточні рухи), епілептичні напади, неконтрольовані напади сміху, відсутність мовної діяльності. Незважаючи на різні фенотипові прояви, мікроде-леції у хромосомі 15, які виявляються при цих синдромах, є ідентичними. Патологічні прояви залежать від того, від кого дитина отримала змінену хромосому: якщо хромосома з мікроделецією дісталася від батька, то симптоматика відповідає синдрому Прадера - Віллі, якщо від матері - синдрому Ангельмана. Отже, важливим є не тільки факт успадкування аномальної хромосоми, а також її походження. Таке явище назвали хромосомним імпринтингом, а хвороби, які спадкуються подібно до синдромів Ангельмана та Прадера - Віллі, - хворобами імприн-тингу. Явище імпринтингу пояснюється різною активністю деяких алелів на материнських і батьківських хромосомах унаслідок ефектів епігенетичної спадковості (див. розділ 6). Таким чином, для генів, котрі підлягають імпринтингу, спостерігається функціональна моносомія, а організми за цими генами є функціональними гемізиготами.
Окремою групою серед патологічних станів, зумовлених генетично, стоять патології несумісності матері та плоду. Найвідомішим прикладом такої патології є резус-конфлікт. У випадку, коли мати резус-негативна (відсутній Rh-антиген), а плід - резус-позитивний, в організмі матері утворюються антитіла проти Rh-антигену. Перша вагітність може пройти без будь-яких проблем. Під час другої вагітності, коли знову розвивається плід із позитивним резус-фактором, виникає конфлікт сумісності між організмом матері та плоду, оскільки мати вже імунна до плоду з позитивним резус-фактором. Цей конфлікт може привести до переривання вагітності або до народження дитини з патологіями.
ГЕНЕТИКА ОНКОЛОГІЧНИХ ЗАХВОРЮВАНЬ
Під онкологічними хворобами розуміють велику групу захворювань (понад 200), які зумовлені появою в організмі змінених (трансформованих) соматичних клітин. Такі клітини характеризуються низкою біологічних особливостей: безконтрольним поділом, безсмертям, порушенням диференціації, здатністю до інфільтрації та знищення сусідніх нормальних клітин, можливістю мігрувати по організму й утворювати метастази - вторинні колонії в органах і тканинах. Розвиток хвороби (канцерогенез) є довгим процесом, усі етапи якого (від ініціації до утворення метастазів) пов'язані з перебудовами геному трансформованих клітин. Найчастіше всі популяції трансформованих клітин в організмі є нащадками однієї зміненої соматичної клітини. Отже, онкологічні захворювання є одним із типів спадкових хвороб - спадкових хвороб соматичних клітин.
Зміни геному, які приводять до трансформації клітин, стосуються генів, відповідальних за регуляцію клітинного циклу, диференціації та запрограмованої загибелі клітин. Крім мутацій, у трансформованих клітинах відбувається перерозподіл епігенетичних маркерів, що викликає зміни експресії деяких генів. Гени, залучені до канцерогенезу, зазвичай поділяють на три групи:
1. Протоонкогени (прийнято позначати c-onc) - гени, нормальна функція яких полягає у стимуляції поділу клітини. До цієї групи відносять також гени білків, що інгібують апоптоз, стимулюють ангіоге-нез (проростання судин в орган чи тканину при недостатності поживних факторів або кисню) і рухливість клітин. Мутації цих генів або епігенетичні зміни, які призводять до їхньої гіперекспресії, стимулюють розвиток новоутворень (протоонкогени перетворюються на онкогени). Мутації, як правило, є домінантними - достатньо однієї події для стимуляції відповідного етапу канцерогенезу.
2. Антионкогени (або гени-супресори пухлин) - гени, нормальна функція яких полягає в затриманні поділу клітин, активації процесів диференціації, стимуляції апоптозу та інгібуванні ангіогенезу. Для розвитку новоутворення в більшості випадків потрібна інактивація обох алелів генів-супресорів (двоударний механізм), тобто онкогенні мутації цих генів є рецесивними.
3. Гени-мутатори (genome care keeper genes - "охоронці геному") -гени, нормальна функція яких полягає в підтриманні цілісності геному (наприклад, гени білків репараційних систем). Їхня інактивація унаслідок мутацій або епігенетичних змін є причиною збільшення частоти будь-яких мутації будь-яких генів - у тому числі протоонкогенів і генів-супресорів. У результаті клітина набуває так званого мутатор-ного фенотипу. Мутації генів-мутаторів є рецесивними.
Вважається, що надбання клітиною мутаторного фенотипу часто є ключовим етапом канцерогенезу. Мутації або інактивація генів білків репараційних систем значно збільшують імовірність виникнення інших онкогенних мутацій, що прискорює проходження всіх етапів канцерогенезу.
Причинами розвитку новоутворень, подібно до інших захворювань, є як фактори навколишнього середовища, так і генетична схильність до хвороби. Близько 80 % усіх випадків онкозахворювань відносять до спорадичних (випадкових), 15 % - до сімейних форм (важко розділити вплив середовища та генотипу), і тільки 5 % суто спадкові.
До середовищних факторів, що індукують розвиток новоутворювань (канцерогенів), належать будь-які мутагенні чинники (див. розділ 4) та онкогенні віруси. Вони трансформують заражену клітину за рахунок онкогенів, які присутні в геномі вірусу. Вірусні онкогени (позначаються як v-onc) можуть бути клітинного походження (захоплений вірусом c-onc), як у багатьох онкогенних ретровірусів, або бути вірусними генами, що кодують білки, необхідні для життєдіяльності вірусу (ДНК-онковіруси: папіломавіруси, вірус Епштейна - Барра тощо).
Середовищні чинники самі по собі відповідають за розвиток невеликої частини онкозахворювань. Розвиток більшості новоутворень (навіть спорадичних) залежить від спадкової схильності. Але на відміну від багатьох розглянутих вище спадкових хвороб, спадкові ракові синдроми мають ряд особливостей прояву, які утруднюють їхнє діагностування. Якщо при інших спадкових синдромах комплекс симптомів є результатом плейотропної дії відповідного мутантного гена, то успадковані онкогенні мутації - тільки "ініціатори" новоутворення. Подальший розвиток хвороби потребує додаткових змін геному окремих соматичних клітин.
Для багатьох спадкових ракових синдромів спостерігається своєрідний парадокс спадкування: на клітинному рівні успадкована мутація є рецесивною (більшість генів, які зумовлюють спадкові ракові синдроми, є генами-супресорами пухлин), а на організменому рівні поводить себе як домінантна. Справа в тому, що будь-яке новоутворення є хворобою соматичних клітин, розвиток якої є випадковою подією. Зрозуміло, що за наявності успадкованої мутації ймовірність ініціації захворювання (мутації та відповідної втрати іншого алеля ге-на-супресора пухлини в будь-якій соматичній клітині, яка в подальшому може дати початок зміненому клону) є на багато порядків вищою, ніж у разі відсутності такої мутації (ймовірність мутації одного алеля становить 10-5-10-7, двох - 10-10-10-14).
Незважаючи на великі труднощі при встановленні спадкової схильності до онкологічних хвороб, розроблено ряд критеріїв, за якими можна з високою імовірністю виявити спадкові форми. Основні ознаки спадкових ракових синдромів такі:
• наявність ідентичних або подібних форм онкозахворювань у двох або більше близьких родичів;
• ранній розвиток (до 45 років) онкозахворювання хоча б у одного з близьких родичів;
• білатеральні пухлини в парних органах;
• велика кількість первинних пухлин у одного хворого;
• домінантний тип спадкування.
Звичайно, для окремих спадкових ракових синдромів існують додаткові критерії підтвердження діагнозу.
Дослідження молекулярних причин ракових синдромів є важливим джерелом інформації про гени, порушення функції яких приводить до ініціації та прогресування онкозахворювання. Популяційний скринінг мутацій цих генів допомагає виявляти групи ризику та проводити більш ефективну профілактику та ранню діагностику онкозахворювань. Вивчення поліморфних варіантів протоонкогенів, генів-супресорів пухлин і генів, які зумовлюють чутливість організму до канцерогенних факторів середовища, дозволяють розкривати причини багатьох онкологічних хвороб і оцінювати індивідуальний ризик розвитку злоякісних новоутворень.
ГЕНЕТИЧНІ АСПЕКТИ ЕВОЛЮЦІЇ ЛЮДИНИ
Реконструкція еволюції біологічного виду Homo sapiens sapiens, як і будь-якого сучасного виду, є важкою для розв'язання проблемою, що залишається далекою від свого остаточного вирішення. Можливі етапи еволюції людини запропоновані на основі палеонтологічних даних, порівнянні варіацій послідовностей ДНК людини та людиноподібних мавп, а також послідовностей ДНК різних груп сучасних і викопних останків стародавніх людей.
За палеонтологічними даними перші гомініди (до гомінід відносяться людина й людиноподібні мавпи) з'явилися в Африці приблизно 20 млн років тому. Чотирма мільйонами років датуються рештки гомінід, які могли би за припущеннями бути предками людини: кеніан-тропів, ардипітеків і австралопітеків. Рід Homo відокремився від стародавніх гомінід приблизно 2,5 млн років тому. Цю групу становили декілька видів, які проживали одночасно.
Із них прямим предком людини, за морфологічними показниками скам'янілих останків, вважається Homo erectus (людина прямоходяча). Вид зародився в Африці, потім представники мігрували в Азію та Європу. За загально прийнятою гіпотезою початок новому виду Homo sapiens у процесі еволюції дали популяції людини прямоходячої, що мешкали в Африці. Вид Homo sapiens мав два підвиди - Homo sapiens neandartaliensis (неандертальці, або палеоантропи) і Homo sapiens sapiens (люди сучасного типу, або неоантропи). Неандертальці з'явилися приблизно 400 тис. років тому, вимерли 30-25 тис. років тому. Сучасні люди виникли приблизно 200 тис. років тому. Деякий час неандертальці й неоантропи проживали на спільних територіях.
Підтвердження та уточнення палеонтологічних даних отримано при порівняльному геномному аналізі сучасних гомінід. Було з'ясовано, що найближчим до людини видом є шимпанзе, а найвіддалені-шим - орангутанг. Порівняння каріотипів шимпанзе й людини показало, що 13 хромосом є повністю ідентичними за морфологією та особливостями диференційного забарвлення. Хромосома 2 людини утворилася в результаті злиття двох акроцентричних хромосом, ідентичних тим, які присутні у хромосомному наборі шимпанзе (саме тому людина має 23 пари хромосом, а шимпанзе та інші людиноподібні мавпи - 24). Решта дев'ять "неідентичних" хромосом відрізняються тільки за наявністю перицентричних інверсій. Така подібність каріотипів характерна для надзвичайно близьких видів.
Більш вражаюча інформація була отримана при порівнянні послідовностей ДНК і білків - генні послідовності людини й шимпанзе збігаються майже на 99 %. Така подібність є зазвичай характерною для "видів-близнюків", тобто таких видів, які морфологічно неможливо відрізнити. Це дало навіть підставу деяким дослідникам запропонувати приєднати шимпанзе до роду Homo. Причиною суттєвих морфологічних відмін між шимпанзе та людиною вважають особливості експресії генів, хоча питання залишається далеким від свого вирішення.
За допомогою порівняння геномів можна встановити не тільки генетичну подібність різних видів, але, використовуючи метод молекулярного годинника, і з'ясувати час, коли відбулося розгалуження еволюційного дерева. Метод молекулярного годинника базується на факті, що в різних ділянках геному мутації з'являються з різною частотою. Знаючи таку частоту (а, відповідно, і середній час, за який виникає одна мутація) для певної геномної зони, можна розрахувати час, потрібний для виникнення певної кількості розбіжностей між геномами, що аналізуються. Для подібних досліджень найбільш активно
використовують послідовності мітохондріальної ДНК і нерекомбіную-чої ділянки Y-хромосоми. Ці послідовності мають дві значні переваги: їхні зміни виникають винятково в результаті мутацій (відсутня рекомбінаційна мінливість), і передача цих послідовностей нащадкам є одно-батьківською (мтДНК передається по материнський лінії, Y-хромосома - по батьківській). Так, раніше вважалося, що відокремлення гілки предків сучасного орангутанга від загального дерева гомінід відбулося приблизно 10 млн років тому, а 5 млн років тому відбулося одночасне розділення гілок людини, шимпанзе та горили. При порівнянні мтДНК людини й людиноподібних мавп було встановлено, що гілки шимпанзе та горили відокремилися значно пізніше, тобто тоді, коли предки людей уже повинні були існувати.
Порівняння мітохондріальної ДНК сучасних людей і неандертальців підтвердили, що ці дві групи є більш генетично близькими, ніж шимпанзе та людина, і належать до одного виду. Загальний предок неандертальців і людей сучасного типу жив приблизно 500 тис. років тому. На сьогоднішній день не має молекулярних доказів того, чи схрещувалися між собою неандертальці та неоантропи.
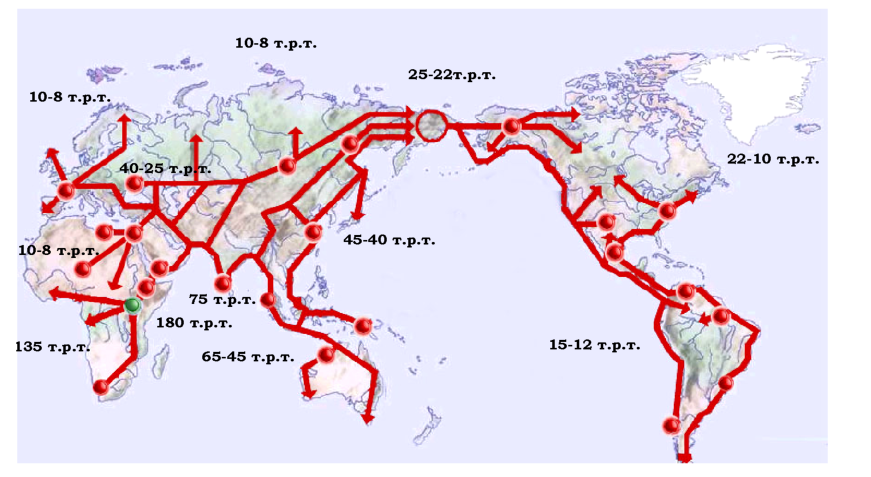 |
|
Рис. 7.8. Шляхи міграції людства за період 180-8 тис. років тому (т.р.т.). Зеленим колом позначений центр зародження сучасної людини. Червоними колами - місця значних палеонтологічних знахідок. (Детальніше див. на http://www.bradshawfoundation.com) |
За даними аналізу ДНК (які підтверджують сучасні палеонтологічні дані) люди сучасного типу виникли приблизно 180 тис. років тому в одному регіоні східної Африки й розселилися по всій земній кулі. Аналіз послідовностей ДНК дозволив також з'ясувати основні шляхи міграцій людства за приблизно 150 тис. років (рис. 7.8). Крім того, порівняння мтДНК і Y-хромосоми показали, що всі відомі варіанти цих послідовностей (як сучасні, так і з викопних останків) виникли в результаті послідовних однонуклеотидних замін, тобто всі ці варіанти можна звести до однієї первинної послідовності. Аналіз мітохонд-ріальної ДНК свідчить, що всі сучасні люди є нащадками однієї жінки (так званої "генетичної Єви"), а аналіз Y-хромосоми, - що предком усього людства був тільки один чоловік ("генетичний Адам"). Слід зауважити, що хоча "прабатьки" всього людства походять зі східної Африки, жили вони в різні часи: генетична Єва жила 160 тис., а генетичний Адам - приблизно 60 тис. років тому.
Існування загальних "прабатьків" усього людства та розбіжність у періодах їхнього життя можна пояснити випадковими причинами: велика кількість ліній, що йшли від інших прабатьків, просто обірвалися. Справа в тому, що декілька разів за свою історію людина як вид була на межі вимирання. Наприклад, вибух вулкану Тоба, який відбувся 74 тис. років тому, ініціював початок льодового періоду. Унаслідок цього катаклізму загальна кількість людей на Землі скоротилася до 10 тис., а саме людство існувало у вигляді розкиданих невеличких ізолятів. Неодноразове проходження людства скрізь таку "шийку пляшки" (див. розділ 8) могло бути причиною того, що випадково залишились нащадки тільки однієї жінки та одного чоловіка.
Контрольні запитання і завдання
1. Охарактеризуйте предмет і завдання генетики людини.
2. Обґрунтуйте недоліки та переваги людини як генетичного об'єкта.
3. Опишіть основні риси організації ядерного геному й каріотипу людини.
4. Перерахуйте типи структурних варіантів геному людини. Як використовується геномний поліморфізм для потреб медичної генетики та судово-медичної експертизи?
5. Як використовується метод складання родоводів при визначенні типів спадкування у людини? Якими основними символами послуговуються при графічному відображенні генеалогічного дерева?
6. Які основні характеристики родоводу вказують на аутосомні типи спадкування ознак? На типи спадкування ознак зчеплених зі статтю?
7. Як залежить прояв багатофакторних ознак від факторів навколишнього середовища? Опишіть модель порогу.
8. Які методи в генетиці людини застосовують для відокремлення дії факторів зовнішнього середовища від впливу генотипу на прояв ознаки? Дайте визначення термінам конкордантність і дискордантність.
9. За даними, наведеними в табл. 7.3, розрахуйте коефіцієнт спадковості для епілепсії.
10. Як можна поділити хвороби залежно від внеску спадкових і середо-вищних факторів у розвиток патологічних станів?
11. Класифікація спадкових захворювань. Наведіть приклади генних спадкових захворювань.
12. Які основні характеристики хромосомних спадкових захворювань?
13. Назвіть трисомії та моносомії, які в людини є життєздатними.
14. Які існують мікроделеційні синдроми та хвороби імпринтингу?
15. Поясніть причини патологічних станів несумісності матері та плоду.
16. Опишіть особливості, які відрізняють онкологічні захворювання від інших спадкових хвороб.
17. В яких клітинних процесах беруть участь протоонкогени, антионко-гени та гени-мутатори?
18. Як саме відрізняються хромосомні набори людини та шимпанзе? Наскільки коректно відносити людину та шимпанзе до "видів-близнюків"?
19. Застосування методу молекулярного годинника при дослідженні еволюції людини. Де й коли, за даними генетичних досліджень, зародилась людина сучасного типу?
20. Чим можна пояснити, що всі сучасні люди мають спільних прабатьків?
Автор: admin от 18-07-2013, 17:02, Переглядів: 35935